Packaging defects still account for the largest proportion of quality deficiencies in medicinal products. The proportion in Germany in recent years has consistently been in excess of 40 %, a figure which the authors of this article consider to be unnecessarily high, and which could be avoided if suitable measures were taken. Based on their experiences in consultancy practice, the authors take examples of three types of packaging – four-side sealed pouches, blister packs with push-through aluminium foil, and plastic closures for glass tablet jars – and show how packaging defects can be recognised, detected and remedied. At least as important as the recognition of packaging defects is the employment of suitable prevention strategies, and these are also explained. If such strategies are to be implemented, familiarity with the packaging process and its limitations, and a comprehensive understanding of the relevant packaging material and the properties of the packaging are essential. In this regard, faultless, regularly checked and continuously adapted packaging specifications are the key to success.
If a pharmacist suspects quality deficiencies, side effects or misuse of a medicine, he or she notifies the competent authorities in accordance with the pharmaceutical regulations, and the Drug Commission of German Pharmacists ("Arzneimittelkommission der Deutschen Apotheker", AMK) in accordance with the commitment to the code of medical ethics. The AMK, in its capacity as a pharmacovigilance centre, collects, evaluates and reports on these notifications. The AMK also informs pharmacists regularly and promptly of newly emerging risks and regulatory measures concerning particular medicinal products.
In 2018 the AMK office received a total of 9,486 spontaneous reports of quality deficiencies and unwanted effects from 4,846 different pharmacists. Out of 6,527 reports of quality deficiencies, the most common were packaging defects, followed by galenic deficiencies, mechanical defects and declaration errors.
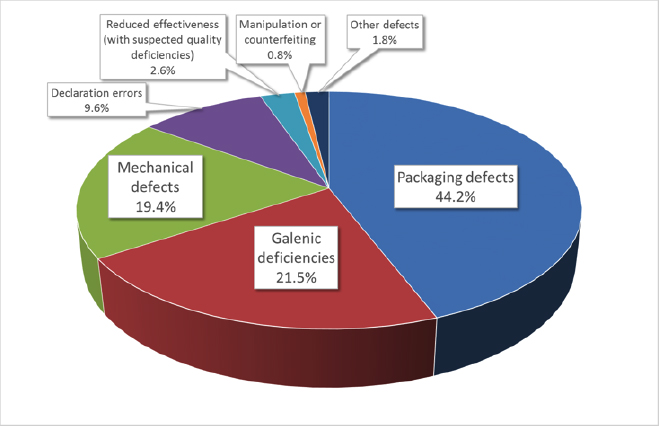
Fig. 1: Suspected cases of quality deficiencies in medicinal products in 2018 (Source of Figs 1 and 2: Prof. Dr. Martin Angerhöfer, based on AMK figures)
In prior years too, packaging defects were the most common cause of quality deficiencies reported to the AMK.
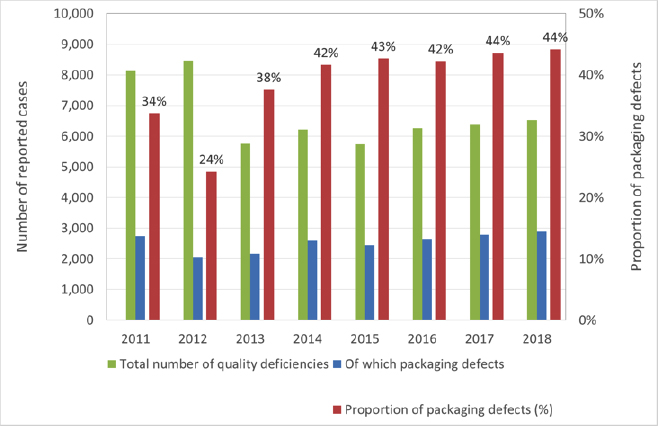 Fig 2: Quality deficiencies and proportion of packaging defects 2011 - 2018
Fig 2: Quality deficiencies and proportion of packaging defects 2011 - 2018
The authors assume that the actual proportion of packaging defects is in fact far greater, because packaging defects that are not immediately recognisable as such can cause deficiencies that would be categorised as “galenic”. One such example, where leaky primary packaging led to discoloration of the medicinal product, will be discussed in greater detail later in this article. A certain number of declaration errors are also directly linked to packaging defects.
As a result, well over half of all reports of deficiencies received from the market are attributable to defects associated with packaging materials or the packaging process as a whole. Similarly, a double-digit percentage of both “Dear Doctor” letters (in German "Rote-Hand-Brief”) and recalls by the German Federal Institute for Drugs and Medical Devices (BfArM) and the Paul-Ehrlich-Institute (PEI) relate to packaging defects.
The examples quoted in this paper are taken from consultancy experience, and serve to illustrate the specific problems associated with the packaging of pharmaceutical products. They are not intended to explain the individual cases down to the very last detail. No mention is made of product descriptions or company names, because similar, entirely unrelated, examples may well crop up again now or in the future. As these types of packaging are used by many different companies, our intention is to draw attention to possible weak points and thus prevent the recurrence of such defects.
Example 1: Four-side sealed pouches
This first example shows the type of packaging known as “multilayer four-side aluminium composite foil” of the sort widely used for powders, tablets and effervescent tablets.
Fig. 3: An example of a 4-side sealed pouch (Source: Romaco Group, Karlsruhe)
The most common type of film structure (from inside to outside) is: HD-PE as a sealant layer, aluminium foil as a barrier layer and a layer of paper for printing. This form of packaging has also been widely used for many years outside the pharmaceutical industry, and is generally mass-produced on high-performance machines. Given the wealth of experience with this type of packaging, problems should be a rare occurrence. However, in cases where the obligatory blue dye test reveals leaky packaging even if the foil specification, machine settings and environment are unchanged, the cause of the fault must be determined as a matter of urgency. In this particular instance, the problem was caused by microscopic holes near the sealing edge in the machine direction, occurring only sporadically and not clearly attributable to any single cause. The initial approach in such cases is always to check the slitting chart, in order to narrow the cause of the problem down to particular production batches or particular rolls. Further checks are carried out on retention samples, the composite films and the individual elements of the composites. If the slitting chart reveals significant defects, and if no serviceable retention samples of the rolls used (aluminium and paper) are available, it becomes extremely difficult to locate and identify the cause of the problem. The composite material for this type of packaging is manufactured by extruding a layer of PE between an aluminium foil and a paper layer (taken from different production batches). A further layer of plastic is applied on another extrusion unit. The result is a continuous composite roll which is then wound round several parent rollers and cut at the end of the process. In the next stage of the process the composite material passes through another machine where it is printed with different designs for different customers and then cut into smaller rolls in accordance with their orders. This production process results in a manufacturing and supply basis which is not directly traceable without an accurate slitting chart. Given this situation, the fault can only be traced by checking the finished composite film batches in the hope of finding deviations from the specifications or from earlier batches that were produced without any problems. In this particular case the very complex search for faults or deviations was successful. The defect was found to be caused by the inadequately annealed aluminium foil batch, which led to the formation of cracks in the composite due to its poor stretching properties.
What generally valid conclusions can be drawn from this example? First, that even in the case of long-standing business relationships, continuous, intensive checking by means of quality audits is essential. All materials supplied on rolls must be clearly and unambiguously identifiable by means of a slitting chart. One way of checking would be to select from the store a roll that had already been delivered, and classify it as “defective”. Then, together with the supplier, to try and trace the semi-finished rolls, right back to the parent roll. A sufficient quantity and quality of retention samples taken from the primary materials used in the parent roll must be available in store for the purposes of making these subsequent checks. The same obviously also applies to all the intermediate stages. Material specifications that have been used for many years must be checked at regular intervals (e.g. every 3 years), and if necessary revised, even if no changes have been reported. Clearly this also applies to all the specifications of the upstream supplier. The pharmaceutical customer must always be notified in advance of, and give his approval to, any change of supplier or revisions to the specification. This also includes any changes in nomenclature, even if these are simply the result of IT modifications. Any reference to data sheets or standards must always mention their date of creation or issue, because extremely important details contained in such documents may have been altered, and these could lead to serious misinterpretations in the case of a change of supplier, for example. Any variations in the usual production process must be documented in the delivery documents and if necessary discussed in advance with the customer. All the principles mentioned here must be clearly and bindingly specified. Together with the other order documents they constitute the legal basis for the supply of packaging materials.
With this last example, irrespective of the product to be packaged, a check must be carried out to determine whether the composite film has been stretched. If so, the amount of stretching must be specified with a sufficient degree of accuracy. The vacuum testing carried out as a form of in-process control using a blue dye test only reveals liquid leakages. In the case just described, the extent of the defect was considerably greater, because although the pouches were manufactured with an intact and liquid-tight plastic film, the aluminium foil was torn.
Example 2: Aluminium foil blister pack
This second example also demonstrates that the use of the blue dye leak test as a means of in-process control has limited suitability for the detection of defective packaging. Blister packs for solid dosage forms are probably the most common form of packaging used in the pharmaceutical industry. They comprise a flat film of plastic (PVC, PVDC, PP, PET or a composite) or aluminium which is thermoformed on a packaging machine, filled and then sealed with another flat film, usually aluminium (or in very rare cases mono-PP film). Normally the gas permeability of the thermoformed plastic film determines the imperviousness of the packaging and thus the shelf life of the product. This technology has been used for many years and is generally problem-free. However, if a shelf life test carried out six months after packaging shows that several batches of moisture-sensitive tablets are completely outside the specifications, and a check on the blister packaging reveals no evidence of defects, then we are confronted with a sizeable problem. In this case too, the parameters for all the batches remained unchanged, but the results differed. The first step was to examine the production process in chronological order with a view to establishing whether the logbook entries revealed any packaging machinery downtime. The results revealed that all the defective batches had been manufactured sequentially, so the extent of the defect could be narrowed down to just a few batches. Detailed light-microscopic analysis at large magnification showed that the structure of the seal was not consistent, and differed between the “good” and “bad” batches. Using a method devised at Munich University of Applied Sciences, the thermoformed film was separated from the covering foil. When placed on a light table it was possible to see evenly distributed pinholes over the entire blister pack.

Fig. 4: Holes in the aluminium foil revealed in transmitted light (Source of Figs 4, 6 and 7: Prof. Dr. Martin Angerhöfer)
The fracture sites in the aluminium covering foil were made even more clearly visible using microtome sectioning.

Fig. 5: Mikroskop XL = XL microscope; Detektor BSE = BSE detector; Vergrößerung = Magnification; Beschleunigungsspannung = Acceleration voltage
Fig. 4 shows the separated aluminium foil in transmitted light. The black areas show where the foil is undamaged. The light areas indicate ruptures in the foil, in this case affecting the entire sealing area. The barrier function of the aluminium foil is thus rendered largely ineffective.
The cross-sectional view in Fig. 5 clearly shows the holes. As this is an aluminium thermoformed blister pack, the lower, undamaged layer (light grey) is the aluminium foil. Over a relatively short period of time water vapour was able to pass through the seal and come into direct contact with each individual tablet.
This defect was caused by an incorrectly mounted heat sealing plate. Subsequent technical modifications to the heat sealing station eliminated the problem. As a further safeguard the blister packs were examined visually using a high-resolution magnifying glass. Employees in production and quality control received appropriate training and an inspection instruction sheet was drafted.
The important thing is that in-process and approval testing of blister packs should not be based solely on a blue dye leak test. In the case of composite materials in particular (e.g. PVC/PVDC or other plastic or aluminium composites), defects, cracks and the like are difficult, if not impossible to detect, and only come to light months or even years later in the form of an inadequate shelf life. In the case of single-layer materials (PVC, PP, PET), a wall thickness control test can be used to check uniform thermoforming and thus reveal any variations.
During the course of this complaint procedure a completely new test method was developed to determine the sealing cavity depth. Light-microscopic surface topography was used to measure the sealing profile of the blister pack at defined points and the results were evaluated by means of an algorithm. Sample results are shown in Fig. 6 (a defect-free blister pack) and Fig. 7 (a blister pack with defects in the aluminium foil). It should be noted that the axes indicating the thermoforming depths in Fig. 6 and Fig. 7 are differently scaled.
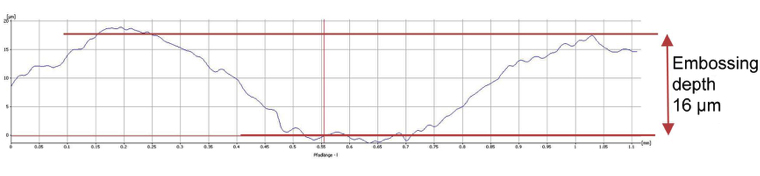
Fig. 6: Topography and embossing depth of a defect-free blister pack (Source: Prof. Dr. Martin Angerhöfer)
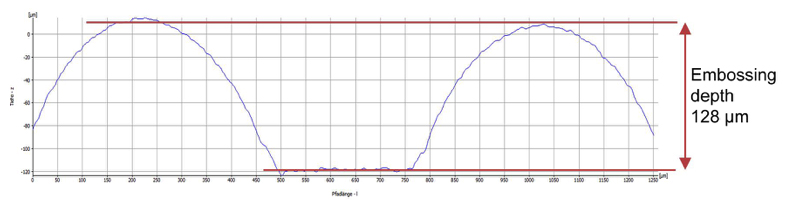
Fig. 7: Topography and depth of a defective blister pack with an embossing depth 8 times greater than the defect-free blister pack) (Source: Prof. Dr. Martin Angerhöfer)
In its present form this method is not yet suitable for in-process control, but it can provide valuable results when it comes to qualifying new machinery/tooling.
Example 3: Plastic closure for a glass tablet jar
Another consultancy case concerned a glass tablet jar with a plastic closure which occasionally did not function as required. This led to a risk of product degradation after the jar was first opened, because the user was unable to reseal the jar properly. In this case the company reacted promptly before any complaints were received from the market. Research and tests carried out at Munich University of Applied Sciences showed that climatic factors, handling and the structural features of the closure could, in the worst case, lead to this sort of problem. The practice of dismantling the injection moulding tool at the end of each production batch in order to clean its individual components created a new configuration when it was reassembled, as a result of which every production batch differed from the previous one. This led to production batches with no in-process faults, and other batches with a higher incidence of defects. Now that the assembly of the moulding tool is carried out systematically and replicably (by labelling the components), the desired improvement has been achieved. In this particular case the company reacted very swiftly to the initial indications of a problem and thus pre-empted any complaints.
With moulds of all types used by packaging material suppliers, the important thing is to know how they are handled during the production of the packaging material and what variations could occur as a result. The service life of moulds should not be judged solely from the point of view of the supplier or in terms of costs. Mould wear can gradually lead to significant functional problems which, if unrecognised, can give rise to complaints and, in the case of crucial functional components, to recalls. If a new mould then needs to be manufactured and qualified a supply shortage lasting several months could result!
Conclusions
These examples show, among other things, that there are a large number of detailed aspects that ultimately decide whether a pharmaceutical product can be consumed/used easily and correctly by the customer. All those responsible for the production of the packaging medium or the packaging material, as well as for the finished medicinal product, must at all times be familiar with their internal processes, and respond accordingly to any deviations. Changes of any kind must be known, and their possible consequences thought through and assessed before they are implemented. Comprehensive knowledge of the manufacturing options and limitations, as well as the properties of the packaging and the packaging material, is essential. The supplier and the customer must work together in a spirit of partnership and openness.
With some consultancy projects it was clear that both the responsibility for and control of quality had been outsourced to the packaging material suppliers, resulting in a long-term loss of know-how on the part of the pharmaceutical customer. If suppliers then cut back on, or largely abandon, quality control due to cost pressures, sooner or later a quality disaster is inevitable.
Faultless, regularly checked and revised packaging material specifications and descriptions are a prime guarantor of success. These specifications must be conceived and drafted in accordance with the customer’s requirements, and must contain technically relevant information for the packaging process. Precursor materials from other manufacturers must be treated in the same manner. The use of manufacturers’ data sheets and generally valid standards is at best a starting point for the correct creation of specifications. Specifications for primary packaging materials should be completely checked and revised at least every three years. In order to counteract “institutional blindness”, this task can be entrusted to external specialists.
When qualifying new packaging machinery or implementing new types of packaging as part of the validation process, it is highly recommended to take every opportunity to test the materials and the packaging. This will provide a verifiable quality standard that can prove invaluable in any subsequent fault tracing, bringing significant savings in both time and costs.